Quantum Computing and Catalyst Design: What Investors Need to Know About Nitrogen Fixation






The molecule that feeds the world, and why it costs so much to make
About half the nitrogen in the human body passed through an industrial reactor at some point. That reactor runs the Haber-Bosch process, the century-old reaction that combines nitrogen and hydrogen to make ammonia, the foundation of synthetic fertiliser. It is, by most measures, the chemical process most responsible for the current scale of human civilisation.
It is also extraordinarily expensive to run. The Haber-Bosch process is responsible for 1–2% of total global energy consumption and 1–3% of global CO₂ emissions, consuming roughly 3–5% of the world's natural gas production each year. To put a number on the scale: around 170 million metric tonnes of ammonia are produced globally each year, with approximately 80% going into fertilisers.
Those figures are not a chemistry trivia problem. They are an investment signal. A meaningful improvement in catalyst efficiency for nitrogen fixation would be one of the largest single decarbonisation and cost-reduction events in the global chemical industry. The question is what it would actually take to find a better catalyst. And that is where quantum computing enters the story.
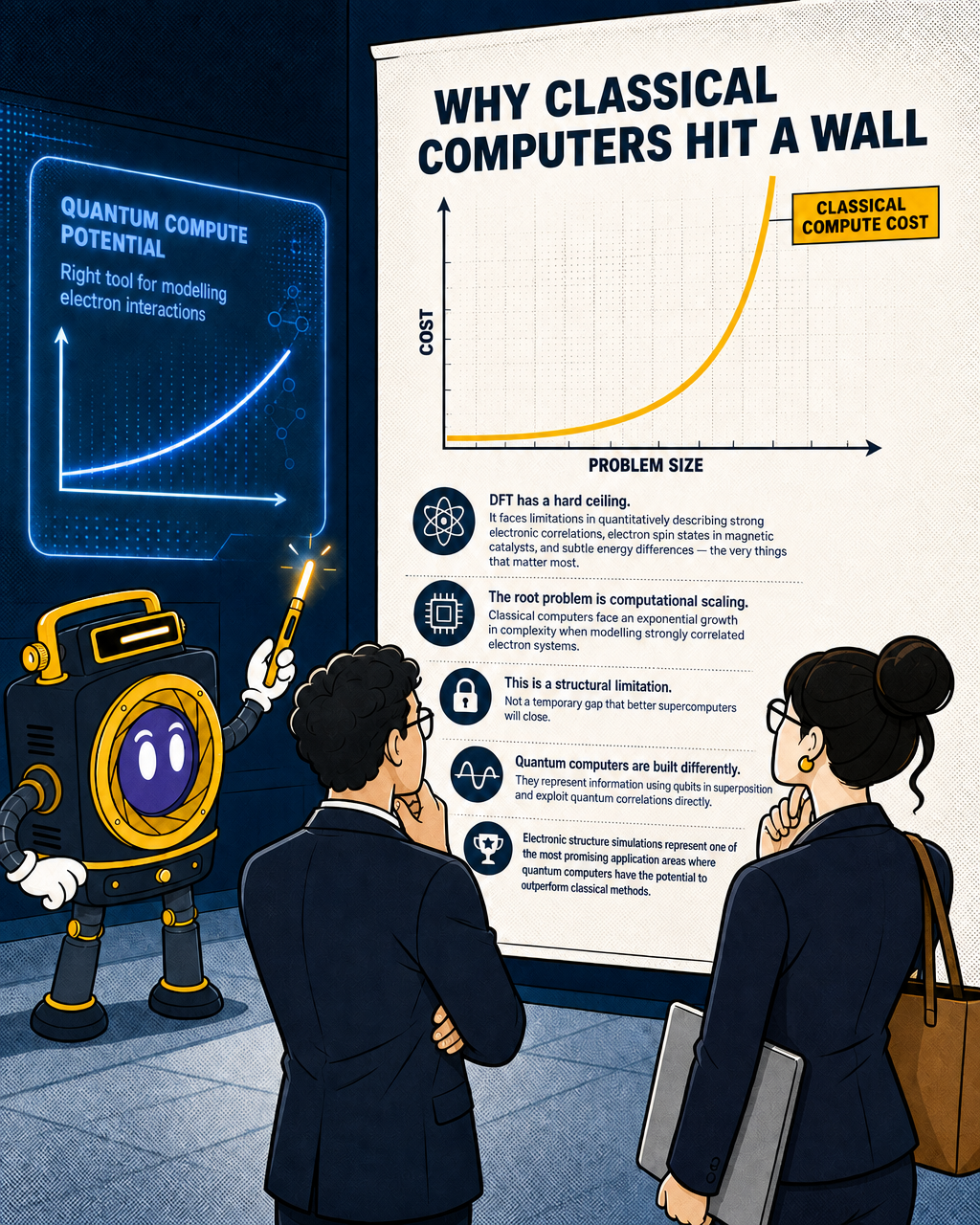
Why classical computers hit a wall
For decades, the standard tool for modelling catalysts computationally has been density functional theory, or DFT. It is a quantum mechanical method that approximates how electrons behave in molecules and materials, and it has been genuinely useful for screening candidate catalysts before running physical experiments.
But DFT has a hard ceiling. It faces limitations in quantitatively describing strong electronic correlations, electron spin states in magnetic catalysts, and subtle energy differences — the very things that matter most when modelling intricate catalytic mechanisms. For most small, well-behaved molecules, DFT is good enough. For the transition-metal active sites where the interesting catalytic chemistry happens, it frequently isn't.
The root problem is computational scaling. Classical computers face an exponential growth in complexity when modelling strongly correlated electron systems, which is precisely the regime where catalytic active sites operate. You can throw more classical compute at the problem, but the cost grows so fast that brute force stops being a viable strategy.
This is not a temporary gap that better supercomputers will close. It is a structural limitation of classical computation when applied to quantum systems.
Quantum computers are built differently. They represent information using quantum bits that can exist in superposition and exploit quantum correlations directly. That makes them, in principle, the right tool for modelling the electron interactions that classical methods approximate poorly. Electronic structure simulations represent one of the most promising application areas where quantum computers have the potential to outperform classical methods.
The molecule that quantum researchers keep coming back to
If you want to understand why the quantum chemistry community is so focused on nitrogen fixation specifically, you need to know about nitrogenase and its FeMo-cofactor.
Nitrogenase is an enzyme found in certain soil bacteria. It does something the Haber-Bosch process requires 400–500°C and 150–300 atmospheres of pressure to replicate: it splits the triple bond in atmospheric nitrogen and converts it into ammonia. It does this under ambient conditions, at room temperature and standard pressure.
The active site that makes this possible is the iron-molybdenum cofactor, FeMoco. It's a cluster of iron, molybdenum, sulphur, and carbon atoms arranged in a way that makes the nitrogen triple bond accessible to chemistry that would otherwise require enormous energy inputs. Understand FeMoco well enough to design a synthetic catalyst that mimics it, and you potentially have the blueprint for a nitrogen fixation process that runs near room temperature.
The problem is that FeMoco is precisely the kind of system classical computers cannot model accurately. FeMoco contains multiple metal centres (iron and molybdenum atoms), making the spin dynamics particularly complex and difficult to simulate. The canonical resource estimate for FeMoco, established by Reiher et al. in a widely cited 2017 PNAS paper, places the required active space at approximately (54 electrons, 54 orbitals) — a configuration that exceeds what is tractable via exact classical methods such as full configuration interaction, and which remains deeply challenging even for approximate classical approaches such as DMRG and FCIQMC. It is not a clean factor-of-two cutoff, but the scaling cost places accurate simulation firmly out of reach of current classical hardware.
That observation is worth sitting with. The most important catalyst design problem in industrial chemistry is, in a precise technical sense, intractable for classical supercomputers using exact methods. It isn't a matter of waiting for a faster processor.
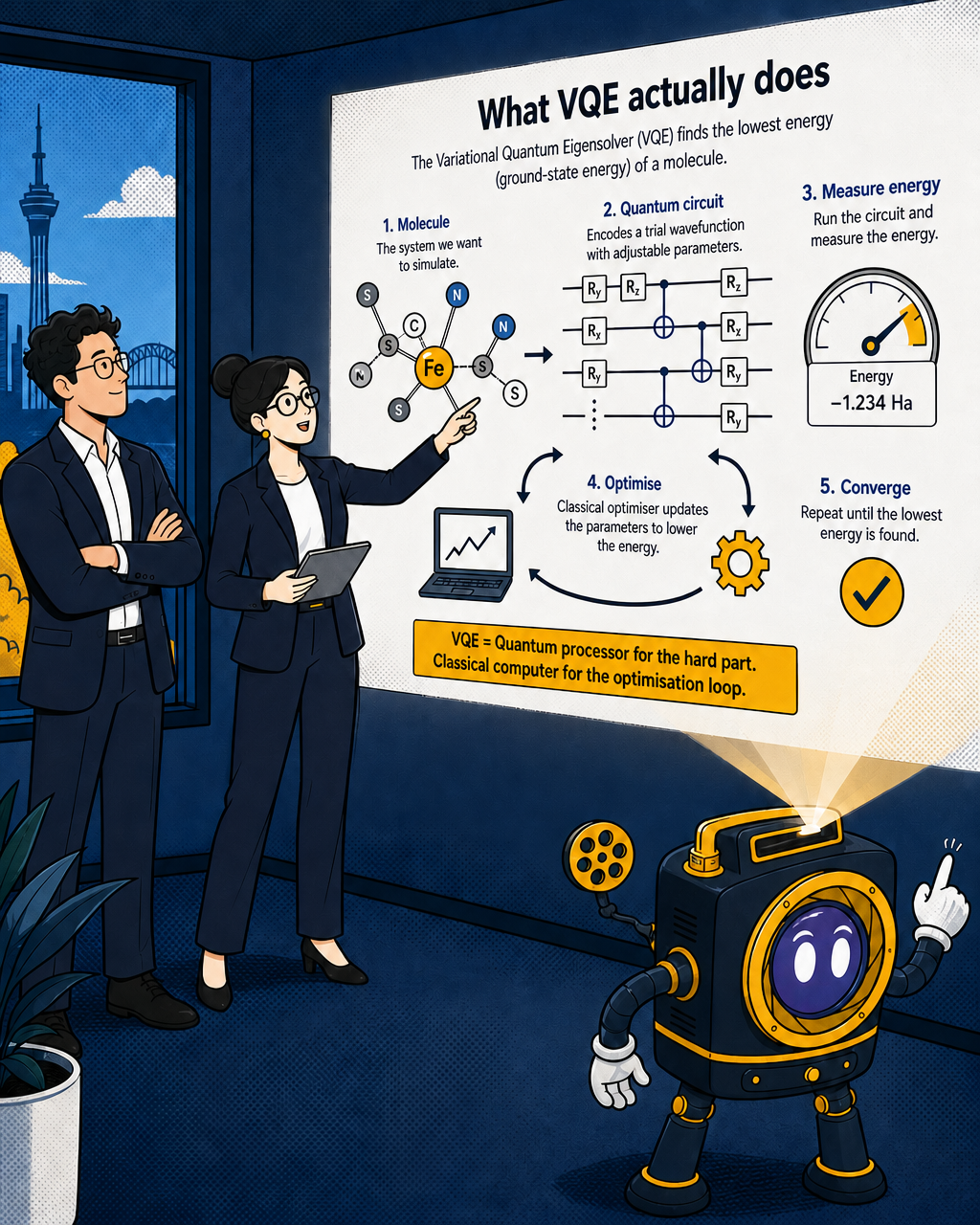
What VQE actually does
The Variational Quantum Eigensolver, or VQE, is the algorithm that has attracted the most attention for this class of problem. It's worth explaining what it does without the jargon.
Every molecule has a ground-state energy: the lowest energy configuration its electrons can occupy, which determines how the molecule behaves chemically. If you can calculate that energy accurately, you can predict how the molecule will react, what bonds it will form, and how efficiently it will catalyse a reaction. For simple molecules, classical methods calculate this well enough. For FeMoco, they don't.
VQE was originally proposed and realised in 2014 on a photonic quantum processor to compute the ground-state energy of a simple two-electron molecule. It has since emerged as a practical tool for calculating ground-state energies in molecular and materials science using quantum computers. The algorithm works by running a quantum circuit that encodes a trial wavefunction, measuring the resulting energy, and iteratively adjusting the circuit parameters until the lowest possible energy is found. Classical optimisation handles the outer loop; the quantum processor handles the inner calculation that classical hardware cannot do well.
For FeMoco specifically, advances in quantum algorithms — including techniques such as tensor hypercontraction and spectral amplification — have progressively reduced resource estimates over the past several years. The Reiher et al. 2017 PNAS estimates were substantially revised downward in Lee et al. 2021 (PRX Quantum), and more recent algorithmic research has continued that trend, with some preprint estimates suggesting requirements on the order of low thousands of logical qubits and hundreds of millions of logical T-gates under specific hardware and compilation assumptions. These figures vary across studies and should be understood as model-dependent projections rather than settled values; resource estimates for FeMoco have historically shifted significantly as algorithmic understanding has improved. Some modelling work suggests that a fault-tolerant quantum computer could simulate FeMoco in hours to days under favourable hardware assumptions, though the actual timeline will depend on clock speeds, error-correction overhead, and gate fidelities of hardware that does not yet exist at scale. The engineering roadmap and the algorithmic requirements are, nonetheless, converging.
The practical implication for catalyst design is significant. A simulation accurate enough to tell you whether a candidate synthetic catalyst will reproduce FeMoco's chemistry would let chemists pre-screen hundreds of molecular architectures computationally before committing to a single physical synthesis run.
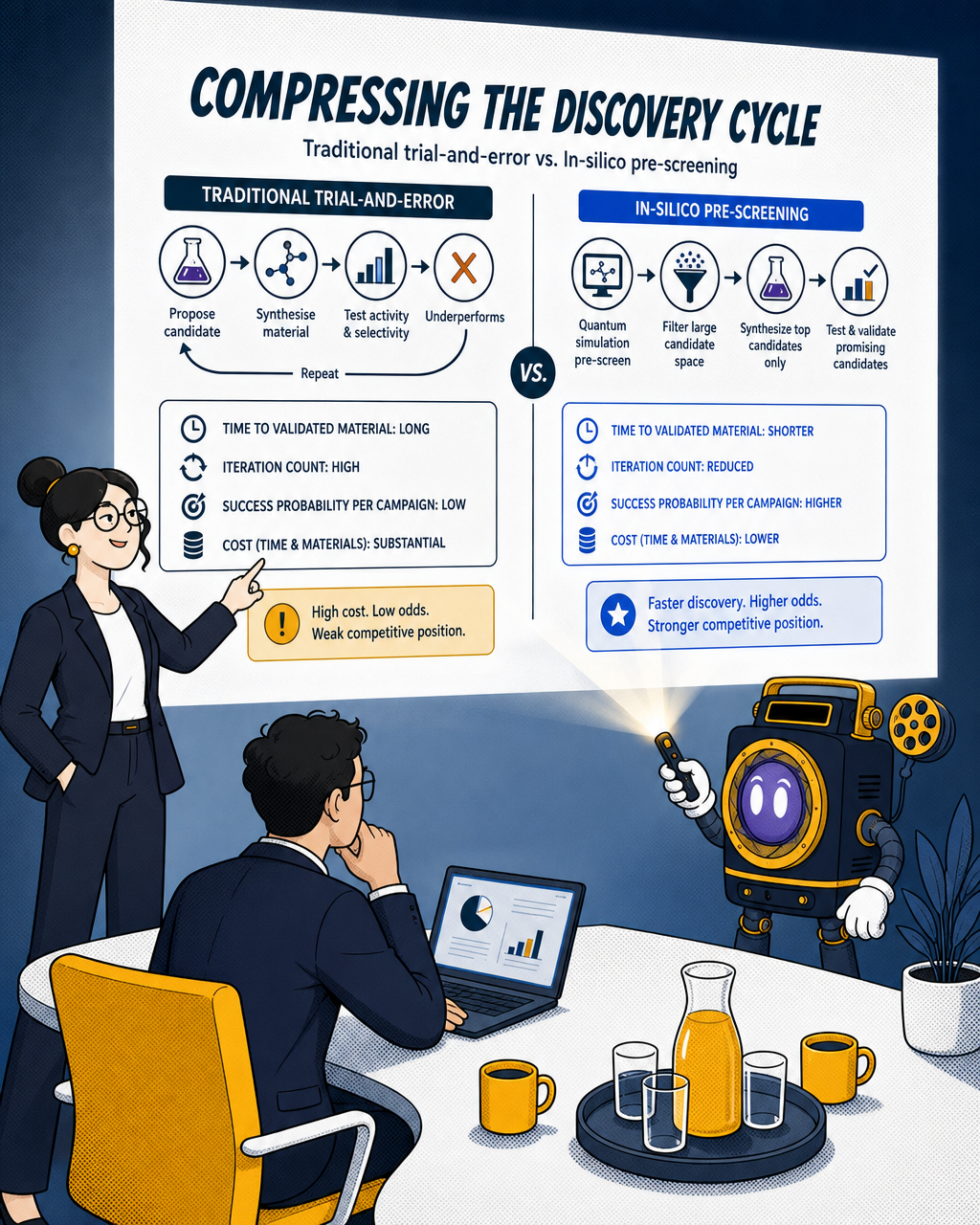
Compressing the discovery cycle
Catalyst discovery today is largely a trial-and-error process. A research team proposes a candidate material, synthesises it, tests its activity and selectivity, finds it underperforms, and repeats. Despite decades of progress through classical molecular modelling methods, catalyst discovery still largely depends on experimental methods with limited theoretical guidance.
The cost of that cycle, measured in time and materials, is substantial. More important for investors is what it means for the competitive position of a company trying to commercialise a novel catalyst: the time from idea to validated material is long, the iteration count is high, and the probability of finding the right candidate in any given experimental campaign is low.
In-silico pre-screening changes that ratio. If a quantum simulation can accurately predict which candidate materials are worth synthesising, teams can filter a large candidate space computationally and only run physical experiments on the most promising subset. Homogeneous catalysis is considered an ideal candidate for demonstrating quantum advantage, with the potential to significantly enhance the capability to design and understand catalytic processes.
The precision required here is non-trivial. For catalyst screening, you don't just need an approximate energy; you need to correctly predict reaction barriers, spin state transitions, and binding energies for specific molecular configurations. Strong electronic correlation, electron spin states, and weak intermolecular forces all present challenges for standard classical simulation methods — and these are precisely the properties that determine whether a catalyst works. Quantum simulation addresses the accuracy gap that makes classical pre-screening unreliable for transition-metal catalysts.
Recent quantum-classical hybrid research has explored modest active-space calculations on current hardware as a step toward practical workflows. Some studies have demonstrated partial correlation energy recovery on small active spaces using limited qubit counts, providing early evidence that hybrid workflows can run on near-term devices — though it is worth noting that recovering a majority of correlation energy is a feasibility demonstration rather than sufficient accuracy for catalyst design decisions, which typically require recovery of 99% or more of correlation energy to achieve reliable reaction barrier predictions. These aren't claims of a solved problem; they are early waypoints on a longer roadmap.
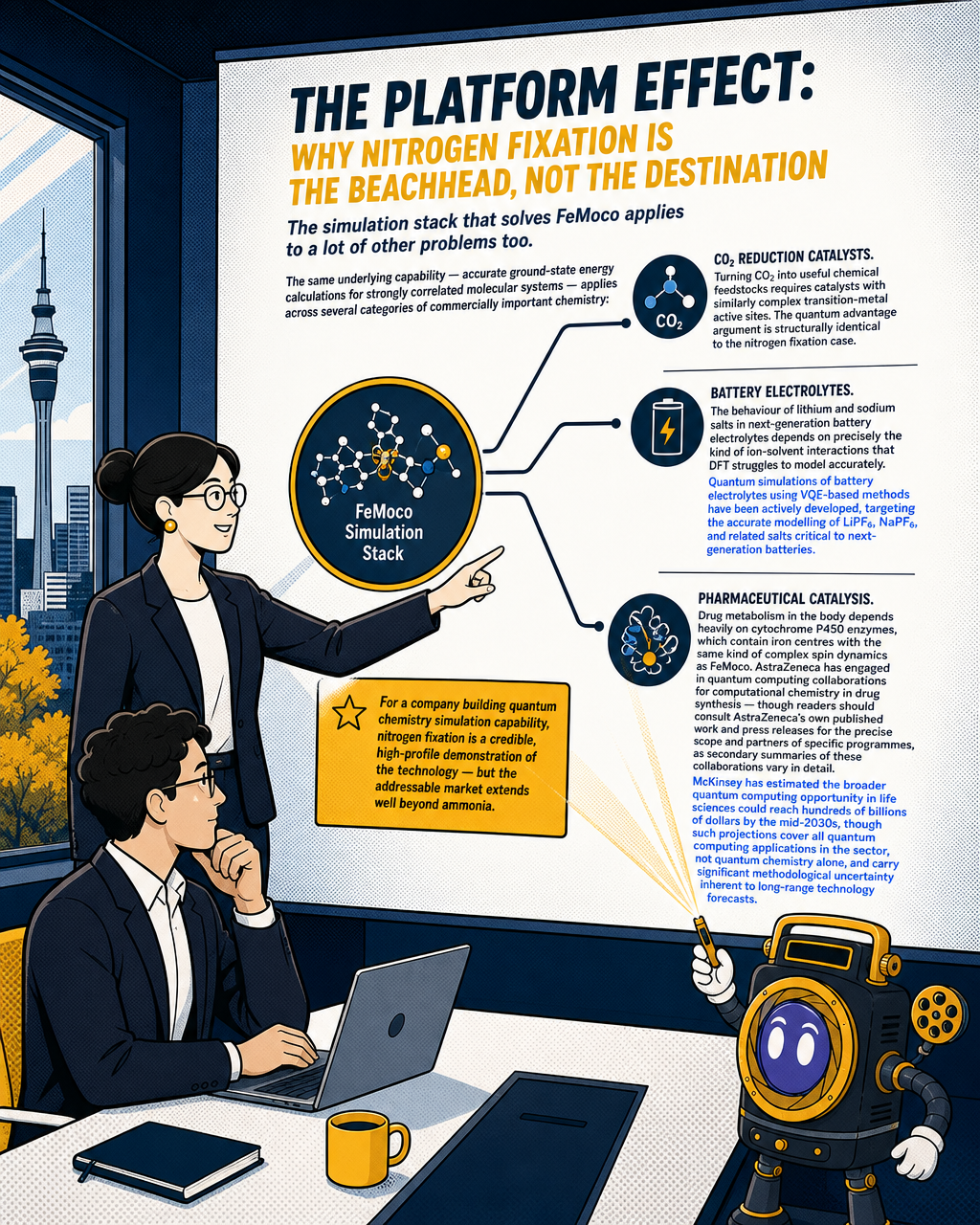
The platform effect: why nitrogen fixation is the beachhead, not the destination
The simulation stack that solves FeMoco applies to a lot of other problems too.
The same underlying capability — accurate ground-state energy calculations for strongly correlated molecular systems — applies across several categories of commercially important chemistry:
CO₂ reduction catalysts. Turning CO₂ into useful chemical feedstocks requires catalysts with similarly complex transition-metal active sites. The quantum advantage argument is structurally identical to the nitrogen fixation case.
Battery electrolytes. The behaviour of lithium and sodium salts in next-generation battery electrolytes depends on precisely the kind of ion-solvent interactions that DFT struggles to model accurately. Quantum simulations of battery electrolytes using VQE-based methods have been actively developed, targeting the accurate modelling of LiPF₆, NaPF₆, and related salts critical to next-generation batteries.
Pharmaceutical catalysis. Drug metabolism in the body depends heavily on cytochrome P450 enzymes, which contain iron centres with the same kind of complex spin dynamics as FeMoco. AstraZeneca has engaged in quantum computing collaborations for computational chemistry in drug synthesis — though readers should consult AstraZeneca's own published work and press releases for the precise scope and partners of specific programmes, as secondary summaries of these collaborations vary in detail. McKinsey has estimated the broader quantum computing opportunity in life sciences could reach hundreds of billions of dollars by the mid-2030s, though such projections cover all quantum computing applications in the sector, not quantum chemistry alone, and carry significant methodological uncertainty inherent to long-range technology forecasts.
For a company building quantum chemistry simulation capability, nitrogen fixation is a credible, high-profile demonstration of the technology — but the addressable market extends well beyond ammonia.
What the timeline actually looks like
It's worth being honest about where the technology sits in June 2026.
Fully fault-tolerant quantum computers capable of simulating FeMoco at the precision needed for definitive catalyst design don't yet exist in commercially accessible form. The algorithms are well-defined. The hardware roadmaps from major players are credible. The resource estimates have improved substantially over the past three years. But near-term quantum hardware still operates with error rates that limit circuit depth.
What does exist, and what is genuinely useful now, is the hybrid quantum-classical approach: using quantum processors to handle the strongly correlated regions of a molecular simulation while classical algorithms address the remainder. This division of labour is already producing results on real molecules. Recent research has demonstrated hybrid methods — combining techniques such as Density Matrix Embedding Theory with quantum diagonalization — achieving agreement with classical reference benchmarks within chemically relevant thresholds for small test systems on current-generation hardware. It is important to note that agreement with a classical benchmark is not the same as agreement with experimental truth; classical methods themselves carry errors that can compound. Nonetheless, demonstrating that quantum-classical hybrid workflows can reproduce established reference results is a meaningful step toward practical utility.
For investors evaluating companies in this space, the relevant questions are: which organisations have built simulation software stacks that will run on fault-tolerant hardware when it becomes available? Which teams understand the chemistry well enough to know which molecular targets are worth solving? And which have built the hybrid workflows that deliver value on today's hardware while the full capability matures?
The comprehension gap in quantum chemistry investment
One pattern that comes up often when quantum chemistry companies try to communicate their value to investors is the gap between what the science means and what the investment case sounds like when compressed into a pitch.
"Quantum simulation of molecular active sites" is accurate but opaque. "We're designing better fertiliser catalysts" undersells the technology. "We're building the molecular simulation infrastructure that could reduce global industrial energy consumption by improving the most energy-intensive chemical process on earth, while applying the same stack to battery electrolytes, CO₂ reduction, and pharmaceutical catalysis" is closer to accurate, but takes about forty-five seconds to say and loses most rooms before the verb arrives.
That comprehension gap is a real commercial problem for companies operating in this space. It isn't a reflection of the quality of the science. It's a reflection of how hard it is to compress a decade of quantum chemistry research into the thirty seconds of clarity that investors, partners, and enterprise buyers actually have available.
The companies that solve the comprehension problem alongside the chemistry problem are the ones most likely to close their rounds, land their commercial partnerships, and build the market position the technology warrants.
If you're building in quantum computing, deep tech, or any sector where the product explanation is as hard as the product itself, Infrairis works with ANZ tech companies to turn complex technical narratives into clear 60-second explainers — delivered in 2–3 weeks, directed by someone who has actually shipped a tech product.
Infrairis
Your complex product. In 60 seconds. Clearly.
Your complex product. In 60 seconds. Clearly.
Learn more about Infrairis and get started today.
Visit Infrairis




